Research Interests
Summary
Dr. Mayfield's lab works on a variety of algae biotechnology research including developing genetic tools and investigating algae as a platform for biofuel and therapeutic protein production. Members of the lab are working on antibody-based therapeutics from recombinant proteins produced from the green algae Chlamydomonas reinhardtii. Other lab members are demonstrating that C. Reinhardtii can be used to produce novel hydrocarbons for the production of superior biofuels. Additionally, the lab is exploring RNA-protein interactions in the chloroplast, creating molecular and genetic engineering tools for algae, and developing high-throughput technology for identifying particular strains with desirable traits.
Expression of Therapeutic Proteins in Eukaryotic Algae
Antibody-based therapeutics have proven to be very effective at treating a number of human diseases, and continue to be one of the fastest growing sectors of drug development. Such therapeutics have great potential fore the treatment of a number of cancers, and offer one of the few options available for treatment of acute infections.
Using the unicellular eukaryotic alga Chlamydomonas reinhardtii, we have developed a system for expression of recombinant proteins. Algae are an ideal system for the production of therapeutic proteins as transgenic algae can be generated in a short time, and the cost of proteins produced in algae is a fraction of the cost of proteins produced by using traditional mammalian cell cultures. Currently, monoclonal antibodies are produced by culturing mammalian cells, and capital costs for production facilities can run into the hundreds of millions of dollars. Because of these high capital and media costs, monoclonal antibodies are some of the most expensive drugs on the market.
To develop an efficient algal-based expression system, we have constructed strains of C. reinhardtii that express a number of variants of human monoclonal antibodies. We have produced several forms of an antibody to herpes simplex virus and have shown that these antibodies assemble in the algae to form fully functional molecules that bind herpes simplex proteins (Mayfield et al, 2003). We have produced antibodies specific for the CD19 protein of lymphoma tumors, and we are exploring the ability of these antibodies to bind and neutralize CD19-positive tumor cells. Recently, we generated a series of transgenic algae that express antibodies to the microorganism that causes anthrax, and have shown that chloroplast expressed anti-anthrax toxin antibodies are capable of biding the toxin at similar levels as mammalian expressed antibodies. We hope to test these molecules for their ability to neutralize anthrax toxin in animal models in the near future.
We have also produced a number of other types of therapeutic proteins, including serum amyloids and human growth hormones. Treatment with serum amyloids may be an effective treatment for the reduction of bacterial and viral infections of the gut, and production of a these proteins in algae makes oral delivery of the therapeutic proteins possible, as green algae are safe to eat. Recently, we achieved expression of therapeutic proteins that accounted for over 5% of total soluble protein produced, levels that make production of proteins in algae economically practical for any type of therapeutic protein (Manuell et al, 2006). Our challenge now is to produce and purify sufficient quantities of therapeutic proteins to begin animal-based testing.
Vector Technology
To truly include Chlamydomonas reinhardtii as a renowned model organism for molecular biology, a wide variety of genetic tools must be developed. In the Mayfield lab, we rely heavily on plasmid vectors for manipulation of the algal genomes. Each vector has been carefully designed by combining specific genetic elements of known function. Many plasmids have been designed with modularity in mind, making it simple to modify a vector to ones needs. This section has been put together to compile a list of our published plasmids as well as to provide a technical resource for those looking to construct their own vectors. Links to purchase a given plasmid through the Chlamy Collection (www.chlamycollection.org) have been provided when applicable.
Chloroplast genome expression
The Chlamydomonas reinhardtii chloroplast is a unique location for protein accumulation. Attributing to its history as an independent prokaryote, the chloroplast contains its own genome with its own unique translational machinery. The chloroplast readily performs homologous recombination, which provides us with a simple means of targeted gene insertion.
Common antibiotics for chloroplast-expressed resistance
- Spectinomycin
- Kanamycin
psbA knockout
Knocking out proteins required for photosynthesis and inserting a heterologous protein sequence has proven to be one of the simpest and most effective ways of accumulating recombinant protein. Most commonly, we use a vector which inserts into the psbA locus.
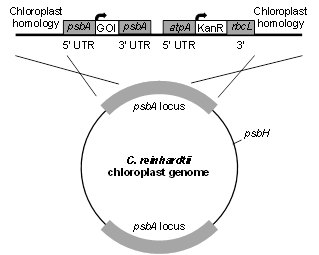 pD1
pD1
This vector uses homologous flanking sequences to replace the endogenous psbA gene with a gene of interest (GOI) and an antibiotic resistance marker. GOI expression and translation is governed by the psbA promoter and UTRs. On the same vector, the AtpA 5’ UTR and rbcL 3’ UTR are used to express and translate either a Kanamycin or Spectinomycin resistance gene. It is important to remember that this construct generates a non-photosynthetic algae, but it can accumulate recombinant proteins at high levels.
Chloroplast silent site integration
When investigating chloroplast promoters, UTRs and regulatory elements, it is important that the vector be integrated to a silent site in the chloroplast genome. Silent site integration generates a photosynthetic strain that can accumulate recombinant proteins at moderate levels.
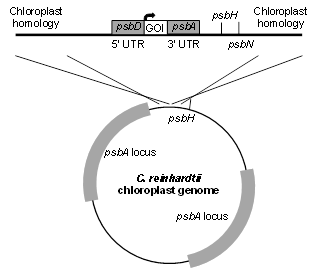 pBR1
pBR1
pBR1 uses homologous flanking sequences to integrate GOI into the 3HB silent site adjacent to the psbH locus. GOI can be governed by any chloroplast promoter and UTR combination. This example shows the psbD promoter and 5’ UTR in combination with the psbA 3’ UTR. This vector is also useful for generating strains in the absence of antibiotic selection. pBR1 can be used to recapitulate photosynthetic competency by replacing an early STOP codon in psbH in the psbH- strain. The selection for transformed algae is photosynthetic competency, thus no antibiotic resistance marker is necessary.
Chloroplast expression resources
Mayfield et al. 2007. Chlamydomonas reinhardtii chloroplasts as protein factories. DOI 10.1016/j.copbio.2007.02.001
Nuclear genome expression
Common antibiotics for nuclear-expressed resistance
- Paromomycin
- Hygromycin
- Zeocin
pBR9: high level nuclear expression
This is the go-to vector for accumulating high levels of recombinant protein through nuclear expression. pBR9 uses the AR1 promoter which is the hsp70A/rbcs2 promoter with the first intron of rbcs2. It also uses the 3’ UTR from rbcs2. pBR9 cleverly utilizes the resistance gene to confer higher protein accumulation. By combining the zeocin resistance gene ble with the foot-and-mouth-disease-virus peptide 2A, we can mitigate nuclear transgene silencing to force the cell to accumulate higher levels of recombinant protein. This vector has been used to express a wide variety of proteins, from the enzymes xylanase and b-glucuronidase to a rainbow of fluorescent proteins.
2A peptide
The Chlamydomonas reinhardtii nuclear genome will often silence transgenes which are not critical to survival. This makes the accumulation of recombinant protein to high levels difficult. To overcome this obstacle, the 2A linker, isolated from the foot-and-mouth-disease-virus (FMDV) can be used to transcriptionally link a gene of interest to a resistance marker, most commonly the zeocin resistance marker ble. The mechanism of ble requires the cell to produce stoichiometric levels of the protein to bind away all of the toxic zeocin. Because resistance to zeocin requires high levels of ble, it upregulates the transcription of the cassette, which also contains the gene of interest. The FMDV 2A sequence is a cys-acting hydrolase element which causes the ribosome to skip during translation and creates two unique peptides; one containing ble-2A and one containing the protein of interest.
Protein targeting using pBR9
In many situations, it is desirable to target recombinant proteins to specific regions of the cell. This technology can be used to help with protein folding or to elucidate organelle mechanisms. In many cases, a simple peptide linker is all that is required to direct the protein to a particular region of the cell. Some of these protein targeting moieties have been investigated by modifying the nuclear expression vector pBR9. Fluorescent proteins and known organelle stains were used to validate the localization.
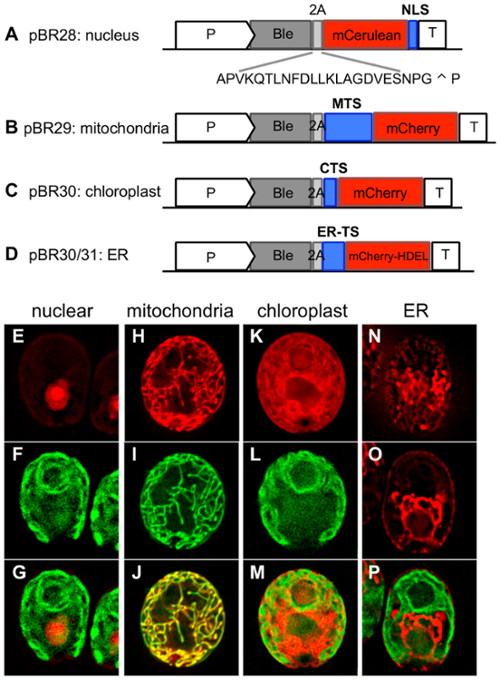 pBR28: Nuclear targeting
pBR28: Nuclear targeting
The nuclear localization signal (NLS) is a tandem copy of the NLS from simian virus 40 (SV40) which can be added to the 3’ end of your peptide for nuclear targeting. This peptide sequence will remain fused to the 3’ end of the protein you wish to target.
pBR29: Mitochondrial targeting
The N-terminal mitochondria transit sequence (MTS) was generated from the nuclear gene encoding the alpha subunit of the mitochondrial ATP synthase located in the mitochondrial matrix. The MTS will remain fused to the 5’ end of a protein of interest and target it to the mitochondria.
pBR32: Chloroplast targeting
pBR32 uses the chloroplast transit sequence (CTS) from psaD, a photosystem I protein. This 35aa sequence was originally used by Fischer and Rochaix in 2001, and has been verified by fluorescent protein localization. The CTS is added to the 5’ end of a gene of interest.
pBR30 and pBR31: ER targeting and localization
The ER is an incredibly important organelle for protein modification and translocation. It connects with the nuclear envelope, houses enzymes used in lipid and carbohydrate metabolism, and acts as a hub for routing proteins for secretion. pBR30 and pBR31 use two different transit sequences to target a protein to the ER. pBR30 uses the ER signal sequence for BIP1 (bipTS), a chaperone of the Hsp70 superfamily which is localized to the ER. pBR31 uses the ER signal sequence for ARS1 (arsTS), a protein which is known to be secreted through the ER. Using either of these signal sequences will target a protein to the ER, but because the ER is particularly competent at mobilizing proteins to other locations, an ER retention signal can be used to keep the protein of interest in the ER. This signal is HDEL (His-Asp-Glu-Leu) and is fused to the C-terminus.
Nuclear expression and targeting resources
Rasala et al. 2012. Robust Expression and Secretion of Xylanase1 in Chlamydomonas reinhardtii by Fusion to a Selection Gene and Processing with the FMDV 2A Peptide. (DOI: 10.1371/journal.pone.0043349) Rasala et al. 2013. Expanding the spectral palette of fluorescent proteins for the green microalga Chlamydomonas reinhardtii. (DOI: 10.1111/tpj.12165) Rasala et al. 2014. Enhanced Genetic Tools for Engineering Multigene Traits into Green Algae (DOI: 10.1371/journal.pone.0094028)
Production of Biofuels in Eukaryotic Algae

Algae offer the potential to produce liquid biofuels at a scale and cost that can be competitive with existing fossil fuel production, and biofuel production from algae will not compete with food crops for arable land use. However, if we are to realize biofuel production from algae in a time frame that coincides with our national needs, we need to immediately begin the development of the molecular and genetic tools that will allow algae to become an economically viable biofuel source.
We also need to train the young scientists whose work will be essential to achieving the production of energy from renewable sources. A key focus of work in my lab is to develop molecular genetic tools in algal while training graduate students and post-doctoral fellows in algal genetics and recombinant technologies, all of which will be essential expertise for producing algae-derived biofuels on a large scale.
With fossil fuel reserves dwindling, mandates requiring the reduction of CO2 emissions, and a need for national energy independence, we face the formidable challenge of developing sustainable forms of carbon-neutral energy in an economically viable manner. Among these new energy forms will surely be biofuels, fuels derived from organisms that capture solar energy for conversion into useable forms of fuel. Two first generation biofuels, ethanol, made by fermenting corn or sugar cane, and biodiesel, made from vegetable oils are already sold in the US. However, the row crops used to produce these biofuels compete with food production and require large amounts of arable land, water and fertilizer, making them less than ideal sources of biofuel. Recently, attention has turned to a promising alternative: single-celled algae. Algae grow quickly, need few added nutrients, and get their energy from sunlight. They can be grown at a very large scale and take CO2 from the air as part of their growth process. Compared with crops normally used to produce vegetable oil such as soybeans or palm, algae can produce 30 times the amount of oil per acre [1]. The US Department of Energy’s Aquatic Species Program - Biodiesel from Algae” highlighted a number of advantages of using algae as a source of biofuels and summarized their finding with the statement “Put quite simply, microalgae are remarkable and efficient biological factories capable of taking a waste (zero-energy) form of carbon (CO2) and converting it into a high density liquid form of energy (natural oil)” [1]. Clearly, algae hold enormous promise to become a major source of renewable energy, especially as transportation fuels, and one that makes sense both economically and environmentally. The challenge in realizing the potential of algae as a biofuel source lies in our relatively limited knowledge of algal physiology and genetics. By developing state of the art molecular genetics tools, and by continuing to develop algal culturing technologies, we hope to optimize algae to be an efficient and economical source of biofuel.
To date, molecular genetic tools have been developed for only a few algae, including the green algae Chlamydomonas reinhardtii, and the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Among these, only C. reinhardtii has well established systems for recombinant protein expression from both the nuclear and chloroplast genomes, and even in this species only a handful of recombinant proteins have been expressed to any appreciable level [2]. Importantly, we have recently been able to demonstrate that C. reinhardtii can be engineered to produce novel hydrocarbon molecules that are superior biofuels, demonstrating the potential of microalgae as a biofuel source. We are developing molecular genetic tools to allow for robust recombinant protein expression in algae, and to engineer algae for the production of biofuel molecules.
Synthetic Biology and Tools for Advanced Algal Engineering
With recent advances in the algal genetic engineering toolkit, we are now able to create complex multi-gene constructs for making large-scale genomic changes in engineered strains. Synthetic biology takes advantage of our ability to create completely novel genetic arrangements to confer desirable traits, with the potential to surpass genetic traits currently found in algal species (see Georgianna and Mayfield 2013 on the Publications page).
For example, we are pioneering methods for designing novel synthetic promoters and untranslated regions (UTRs) to boost transgene expression to levels beyond what is achievable with endogenous regulatory elements. We have used libraries of synthetic oligonucleotides employing mutational scanning across endogenous chloroplast UTRs to identify positive and negative regulatory elements, and then created synthetic UTRs combining multiple positive elements and eliminating all putative negative regulatory elements (see Specht and Mayfield 2013). We are also testing synthetic nuclear promoters derived from computational approaches that identify cis-regulatory architecture in known algal promoters and predict their potential outputs from RNA-seq data.
We are also using a synthetic biology approach to explore the modification of the entire reaction core center of Photosystem II (PSII), to search for novel configurations that increase photosynthetic efficiency or robustness to environmental stresses. We have engineered strains in which all seven core subunits of PSII have been knocked out, and into these strains we can introduce reconstituted photosystems using individual genes from a variety of photosynthetic organisms including cyanobacteria, diatoms, green algae and terrestrial plants (see Gimpel and Mayfield, 2013; Vinyard et al., 2013 and 2014). These experiments have provided valuable insights into the interactions between PSII subunits for functional compatibility, and we continue to explore the potential for this system to achieve enhanced photosynthetic traits.
High-throughput Technology
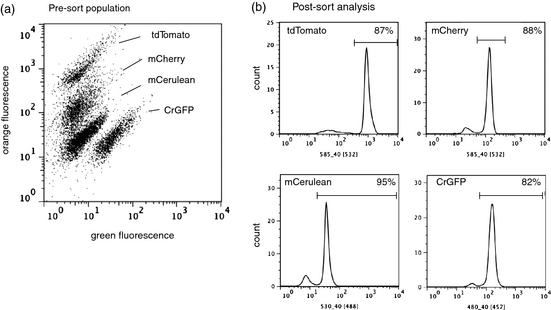
Chlamydomonas reinhardtii is being widely investigated as a production platform for a variety of products. These range from high-value products such as protein therapeutics to large-scale manufacturing of biofuels and nutraceuticals. Before C. reinhardtii can truly become a workhorse for therapeutic protein production, we must identify genomic characteristics which allow the organism to accumulate recombinant protein to higher concentrations. One of the most common methods for generating high levels of recombinant protein currently is by addition of a zeocin antibiotic resistance cassette (Sh ble) to a construct of interest which is inserted into the nuclear genome (1). The mechanism of this resistance gene requires that the cell produce a stoichiometric amount of Sh ble gene product, resulting in high levels of transcription and translation of that gene construct. By coupling this resistance cassette to a gene of interest, one can know that a cell which survives in the presence of zeocin (due to its high levels of Sh ble gene product) will likely contain high levels of recombinant protein.
This process can be time-consuming. For each transformation, cells must be selected for antibiotic resistance, PCR screened for proper introduction of the gene, and western blotted or assayed for recombinant protein activity. This prevents one from screening a high number of individuals for their ability to accumulate the recombinant protein. In reality, the gene is randomly inserted in the nuclear genome and neighboring elements can dramatically change protein accumulation. It is therefore beneficial to screen as many individuals as possible so as to identify the strain with the most advantageous insertion. The Mayfield lab has begun to take advantage of high-throughput screening techniques as a means of selecting these highly-expressing individuals and for identifying the genetic elements which have yielded greater accumulation.
We have developed a rainbow of fluorescent proteins coded for both the nuclear and chloroplast genomes which serve as detectable reporters for fluorescence activated cell sorting (FACS) (2). By using FACS, thousands of cells can be screened every second and cells which have desirable properties can be rapidly isolated and cultured. Figure 1 shows the distinct populations in a 1:1:1:1 mixture of fluorescent protein-containing C. reinhardtii strains. These populations can be sorted cell by cell or in a bulk fashion. There are also dyes available for identifying lipid accumulation, which is imperative for rapid biofuel production optimization.